G Koren1, H Nordeng2 and S MacLeod3
1Motherisk Program, Division of Clinical Pharmacology/Toxicology, SickKids Hospital, University of Toronto, Toronto, Ontario, Canada; 2School of Pharmacy, Oslo University, Oslo, Norway; 3Department of Pediatrics, University of British Columbia, Vancouver, British Columbia, Canada. Correspondence: G Koren ([email protected])
Received 3 October 2012; accepted 20 November 2012; advance online publication 9 January 2013. doi:10.1038/clpt.2012.233
Currently, bioequivalence (BE) studies are carried out exclusively in males, assuming that intrasubject variabilities are similar between the sexes. This article challenges this hypothesis on the basis of available evidence and urges that studies of BE of sufficient power be undertaken in women also for all generic drugs aimed at women.
In the process of introducing a generic drug to the market, its manufacturer is expected, among other requirements, to prove BE to the marketed, reference drug. This is achieved by com- paring systemic exposure, as represented by the area under the concentration–time curve (AUC) of the generic formulation, as compared with the reference drug in a crossover design with the same individuals taking both preparations. To achieve BE, the AUC and peak concentrations of the generic drug need to be within 80–120% of the reference drug.1 These limits are set to ensure comparability in terms of safety and efficacy. Despite US Food and Drug Administration guidelines stating that “if the drug product is intended for use in both sexes, the spon- sor should attempt to include similar proportions of males and females in the study,”1 typically BE studies are conducted in healthy, young adult male volunteers.2
The argument in favor of carrying out BE studies exclusively in males is as follows: even if men and women dispose of drugs differ- ently in terms of absorption, distribution, metabolism, and excre- tion,3 this does not matter because in testing BE, one compares the two preparations using the same individual as his/her own control; hence, being a man or a woman does not matter because the differences between the two formulations, if they exist, will be apparent whether one studies either males or females. This hypo- thetical principle is based on the assumption that intraindividual variabilities in BE are similar between men and women.
The following discussion will examine, based on published data, whether this assumption is supported by evidence and will attempt to answer the question of whether BE of drugs should be tested in women rather than only in men.
AVAILABLE HUMAN DATA
Intrasubject gender variability
In 2000, Chen and colleagues4 from the Center for Drug Evaluation and Research of the Food and Drug Administration reviewed 26 BE studies submitted to the agency in which data involved both men and women. From a total of 23 specific drug studies in which BE was compared between original and generic formulations between women and men, there were statistically different results between genders in five medications (22%) with respect to AUC variability and in four (18%) with respect to variability in peak plasma concentration (Table 1).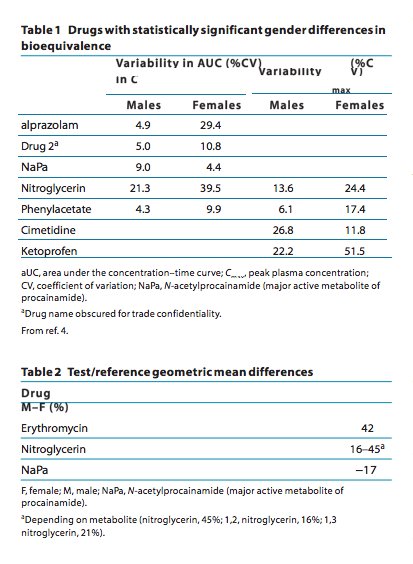 What does this mean practically? These results clearly indicate that, for the drugs listed in Tables 1 and 2, gender difference will have major implications for the conduct and interpretation of BE studies. For example, studies on alprazolam in men sug- gest marginal intrasubject variability, which means that a small number of individuals will be needed to show BE. By contrast, when given to women, the variability in alprazolam jumps six- fold, which means that much a larger number will need to be studied to have the power to document BE. Of importance, in most cases, variability among women is much larger, precluding generalizability from men to women.
What does this mean practically? These results clearly indicate that, for the drugs listed in Tables 1 and 2, gender difference will have major implications for the conduct and interpretation of BE studies. For example, studies on alprazolam in men sug- gest marginal intrasubject variability, which means that a small number of individuals will be needed to show BE. By contrast, when given to women, the variability in alprazolam jumps six- fold, which means that much a larger number will need to be studied to have the power to document BE. Of importance, in most cases, variability among women is much larger, precluding generalizability from men to women.
Of equal importance, there are drugs for which not only the variability in BE is gender dependent but also the BE itself. If one considers erythromycin, BE in men was 42% higher than among women.4 This means that by testing men, a generic drug may be deemed bioequivalent when in fact women would achieve a 42% lower AUC, and the drug would not be approved as interchangeable. Moreover, because different drugs behave differently with respect to gender-specific intraindividual differences in BE, one cannot draw parallels from one drug to another.
Cycle-dependent changes in BE
The fluctuating hormonal status of women along the menstrual period may affect the absorption, distribution, metabolism, and secretion of different drugs. For example, several pharmacokinetic parameters were different for ranitidine according to menstrual period; the AUC was 7,312.15 ng/ml/h in the follicu- lar phase as compared with 5,195.83 ng/ml/h in the luteal phase, whereas in men the AUC was 11,471.94 ng/ml/h.5 It must be concluded that studies of BE of drugs targeting women must compare the reference and generic drug during similar stages in the menstrual cycle.5
Gender-dependent differences in BE due to variability in inactive ingredients
In some cases, adverse drug reactions are due to factors other than the active ingredient in a formulation. Often, a generic drug may differ from the reference formulation in the pres- ence and levels of inactive ingredients. Studying men and women interchangeably in BE studies requires the hypothesis that they handle the inactive ingredients similarly. The fallacy of this assumption has been proven in recent human studies with respect to a common inactive ingredient, polyethylene glycol. Ashiru and colleagues6 have demonstrated that poly- ethylene glycol enhances the bioavailability of ranitidine in a dose-dependent manner by up to 63% among men, whereas it decreases absorption among women by up to 24%!
What do these observations mean in practice? If a ranitidine BE study were to be done in men only, the dose of the generic product, using polyethylene glycol, would have to be 60% lower as compared with the reference drug. If this regimen were then translated to women (as done in almost 100% of studies today), the reduced dose of the generic and the 24% decrease in affect the frequency, onset, and severity of adverse drug reac- tions. The incidence, severity, and duration of adverse reactions and side effects observed during the study must be reported. The probability that an adverse effect is drug induced is to be judged by the investigator. In this context, studies have shown that women may be more sensitive than men to the adverse effects of different inactive ingredients.7,8 Hence, BE studies in men will not necessarily reflect results in women.
DISCUSSION
The consequence of ignoring gender differences in BE studies may result on the one hand in less effectiveness and on the other hand in an increased risk of adverse drug reactions. Although it is well documented that women experience more adverse events than men, and in general these adverse events are of a more seri- ous nature,7,8 we often lack good explanations for these obser- vations. Unfortunately, 8 of the 10 drugs that were withdrawn from the market between 1996 and 2000 were withdrawn due to greater risks of adverse effects in women.9 So why are women not included in BE studies of drugs that will be used also by women?
Compelling evidence has been presented to make the case that one cannot assume BE in women on the basis of studies conducted in men. So why does this practice continue? Why do regulatory agencies still accept results of BE in men for drugs to be used only by women?
Before the twentieth century, men used to perform women’s roles in theater, as a reflection of sexual puritanism. Until recently, women were not allowed to vote in many countries, and this real- ity continues today in some jurisdictions. It was assumed that men knew what was “good for women.” In the case of regulatory reality, the current approach is clearly not good for women, as the science shows. Rather, it reflects the commonly held view that it is easier to study men than women. Women may become pregnant, which is not an issue for short BE studies.6
In reality, BE cannot be deemed similar in men and women for any medication, and, if a generic is to be used also by women, it has to be tested with sufficient power in women.
CONFLICT OF INTEREST
The authors declared no conflict of interest.
Clinical Pharmacology & Therapeutics | VOLUME 93 NUMBER 3 | MaRCH 2013
© 2013 american Society for Clinical Pharmacology and Therapeutics
References:
1. US Food and Drug administration. Guidance for Industry, Bioavailability and Bioequivalence Studies for Orally administered Drug Products -General Considerations. (March 2003, Revision 1) <http://www.fda.gov/downloads/
Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070124. pdf> accessed 31 May 2012.
2. European Medicines agency, Guideline on the investigation of bioequivalence. <http://www.ema.europa.eu/docs/en_GB/document_ library/Scientific_guideline/2010/01/WC500070039.pdf> accessed 3
November 2012.
3. Koren, G. Pharmacokinetics in pregnancy; clinical significance. J. Popul. Ther.
Clin. Pharmacol. 18, e523–e527 (2011).
4. Chen, M.L., Lee, S.C., Ng, M.J., Schuirmann, D.J., Lesko, L.J. & Williams, R.L. Pharmacokinetic analysis of bioequivalence trials: implications for sex-related
issues in clinical pharmacology and biopharmaceutics. Clin. Pharmacol. Ther. 68, 510–521 (2000).
5. Flores Pérez, J. et al. Effects of gender and phase of the menstrual cycle on the kinetics of ranitidine in healthy volunteers. Chronobiol. Int. 20, 485–494 (2003).
6. ashiru, D.a., Patel, R. & Basit, a.W. Polyethylene glycol 400 enhances the bioavailability of a BCS class III drug (ranitidine) in male subjects but not females. Pharm. Res. 25, 2327–2333 (2008).
7. Soldin, O.P., Chung, S.H. & Mattison, D.R. Sex differences in drug disposition. J. Biomed. Biotechnol. 2011, 187103 (2011).
8. Kando, J.C., Yonkers, K.a. & Cole, J.O. Gender as a risk factor for adverse events to medications. Drugs 50, 1–6 (1995).
9. US Government accountability Office (GaO), 2010. Washington, DC, 20548. Drug safety: most drugs withdrawn in recent years had greater health risks for women <http://www.gao.gov/new.items/d01286r.pdf> accessed 31 May 2012.
[...] response to a review of clinical trials on drug bioequivalence, Larry Cahill , PhD, a Professor of Neurobiology and [...]